I have only read the link above, not the paper. This is about the heart's response, not the heart rate. In other words, its about increase in heart rate, not absolute heart rate. I am not sure this explanation is correct, but its the only one that makes sense to me just now. As oxygen use goes up, our heart rate does not go up to match.Why aren't PWME's heart rates going about 60 bpm? I thought our problem was struggling to keep our HRs below our aerobic (or something) thresholds?
-
Notice: S4ME will have a short interruption of service on May 30. More information here.
You are using an out of date browser. It may not display this or other websites correctly.
You should upgrade or use an alternative browser.
You should upgrade or use an alternative browser.
Frontiers review - Chronotropic incompetence an overlooked determinant of symptoms and activity limitation in ME/CFS (prov. 2019) Davenport et al
- Thread starter Ravn
- Start date
Makes so much sense to me, I always feel that my heart can't sustain any simple activity..like I'm in some kind of heart failure. I'm delighted to this study published. Also, HR has nothing to do with pem for me. I can get severe pem from having a visitor.
I have only read the link above, not the paper. This is about the heart's response, not the heart rate. In other words, its about increase in heart rate, not absolute heart rate. I am not sure this explanation is correct, but its the only one that makes sense to me just now. As oxygen use goes up, our heart rate does not go up to match.
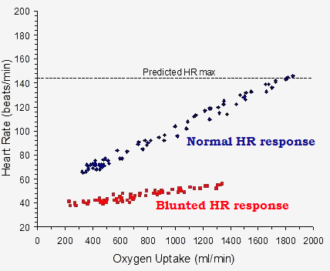
chronotropic incompetence = inability to raise heart rate
Heart rate:
40 - 60 (+50 %) = mecfs
60 - 140 (+230 %) = controls
VO2 oxygen uptake:
200 - 1400 (+ 7x) = mecfs
300 - 1900 (+ 6x) = controls
why didnt they force the mecfs to go to the "predicted heart rate max" ?
what would happen ?
would have thought, that encountering a thing that causes damage to me, would increase heart rate ?
we came in near-coma and left in near-coma... so much for the stepper
now, the "emotional pem".
does watching a horror movie increase your heart rate, after all ?
is it possible to rent such a heart monitor, perhaps in the pharmacy ?
we came in near-coma and left in near-coma... so much for the stepper
now, the "emotional pem".
does watching a horror movie increase your heart rate, after all ?
is it possible to rent such a heart monitor, perhaps in the pharmacy ?
Wonko
Senior Member (Voting Rights)
This makes no sense to me.
I have only experienced a fixed HR during one period, when I was on a beta blocker, atenolol. Then I was restricted because my HR wouldn't go above 60bpm. Given I have traditionally used 'adrenaline' to enable me to get stuff done when I need to not being able to was crippling.
My HR is traditionally high, tachycardic in fact, often even when at rest. Standing can increase my HR to the 120-140 bpm area. An instant of the wrong type of thought can increase it to 160bpm for tens of minutes.
I didn't think I was alone in this amongst PwME, but this suggests I am.
I have only experienced a fixed HR during one period, when I was on a beta blocker, atenolol. Then I was restricted because my HR wouldn't go above 60bpm. Given I have traditionally used 'adrenaline' to enable me to get stuff done when I need to not being able to was crippling.
My HR is traditionally high, tachycardic in fact, often even when at rest. Standing can increase my HR to the 120-140 bpm area. An instant of the wrong type of thought can increase it to 160bpm for tens of minutes.
I didn't think I was alone in this amongst PwME, but this suggests I am.
Wonko
Senior Member (Voting Rights)
If I work really hard at resting then I can get my HR down to high 60s, occasionally, more often low to mid 70s, but I have to be well rested for that, and anything more than a few minutes upright will send it at least into the high 90s. Once it's been like that for a few minutes it rises to low 100s even if I'm not doing anything. If I then do stuff it's 115-130. This is an improvement on how it used to be.
And so it goes.
How long this takes may, but only may, depend on how 'rested' I am, but once my HR is high even lying down immobile doesn't cause it to drop much, for quite a while - in fact it's often higher when lying down after 'exertion' (e.g. standing up long enough to make a cuppa - that sort of 'exertion').
I'm still trying to get a handle on what's going on, but it seems to depend, at least partially, on something I am not monitoring.
I've tried seeing what effects eating, different types of food, not eating, altering the timing and duration of sleep, adding in a few seconds of 'high intensity' exercise (a few reps of a minimum weight deadlift every couple of days), varying durations of 'low intensity' exercise (lurching).
The results make no sense e.g. 3 reps of deadlifts actually seem to reset my HR down to high 70s - low 80's, no matter what it was before, but walking, that raises it, and it stays elevated for anything from 5 hours to days, even whilst sleeping.
It's nonsense.
So the above paper, runs counter to my experience. If it is valid for all PwME then I ain't one, no matter what my symptoms suggest..
And so it goes.
How long this takes may, but only may, depend on how 'rested' I am, but once my HR is high even lying down immobile doesn't cause it to drop much, for quite a while - in fact it's often higher when lying down after 'exertion' (e.g. standing up long enough to make a cuppa - that sort of 'exertion').
I'm still trying to get a handle on what's going on, but it seems to depend, at least partially, on something I am not monitoring.
I've tried seeing what effects eating, different types of food, not eating, altering the timing and duration of sleep, adding in a few seconds of 'high intensity' exercise (a few reps of a minimum weight deadlift every couple of days), varying durations of 'low intensity' exercise (lurching).
The results make no sense e.g. 3 reps of deadlifts actually seem to reset my HR down to high 70s - low 80's, no matter what it was before, but walking, that raises it, and it stays elevated for anything from 5 hours to days, even whilst sleeping.
It's nonsense.
So the above paper, runs counter to my experience. If it is valid for all PwME then I ain't one, no matter what my symptoms suggest..
Mithriel
Senior Member (Voting Rights)
My heart rate goes up when I do anything and can go very high if I am forced to stay upright (doctor's office was worst) but from tracking my HR I have found that when I am particularly bad my heart rate drops. At the same time I can feel dizzy and ill if I try to do something.
I have seen this phenomenon happen to other people in HR monitoring groups so I am not alone.
I have seen this phenomenon happen to other people in HR monitoring groups so I am not alone.
Jonathan Edwards
Senior Member (Voting Rights)
I don't understand the figure. Presumably there are lots of dots, some on top of each other, because there are lots of subjects or test runs. But shouldn't a test run be given by a line - staring at low oxygen uptake and moving to high? And why does the control line start higher for oxygen uptake?
If the x axis is change in heart rate rather than actual heart rate why isn't there much more scatter?
I am obviously not understanding what is being shown.
If the x axis is change in heart rate rather than actual heart rate why isn't there much more scatter?
I am obviously not understanding what is being shown.
Snow Leopard
Senior Member (Voting Rights)
why didnt they force the mecfs to go to the "predicted heart rate max" ?
what would happen ?
If they truly have chonotropic incompetence, there is no "forcing", the patients will simply need to stop early.
What is interesting is that in young healthy people, deconditioning can actually result in slightly higher than normal heart rates at VO2Max than trained participants.
I'm very curious- what could be determined if EMG signalling is measured? On which nerve(s) would the EMG be conducted?
What would an increased latency or reduced rate of HR rise indicate?
The idea is to see if there is something interfering with normal heart rate response to exercise, at least in the subgroup of patients considered to have chronotropic incompetence. Increased latency would suggest the normal mechanisms are not functioning correctly. Likewise, if muscle drive increases but heart rate does not increase appropriately, it suggests there is a problem. This is also assuming "normal" levels of ventilation (no hypoventilation).
An example of a surface EMG study (vastus lateralis in this case):
https://www.ncbi.nlm.nih.gov/pubmed/27697301
An appropriate study of EMG and their interpretation is admittedly a little more complicated than many people in the field suggest if you want to consider the number of muscle fibres activated and want to use this to imply something about the twitch characteristics of those fibres.
https://www.physiology.org/doi/full/10.1152/japplphysiol.00280.2015
Also, for the technically inclined:
https://www.physiology.org/doi/full/10.1152/japplphysiol.00162.2014
Some reading (healthy participants):
"Kinetics of heart rate responses to exercise"
https://www.tandfonline.com/doi/abs/10.1080/02640418808729792
"Early ventilation-heart rate breakpoint during incremental cycling exercise."
https://www.ncbi.nlm.nih.gov/pubmed/23945972
In the above study, note the difference in HR slope before and after the VT, also note that heart rate response to an increased rate of breathing (with a small amount of latency).
Gravier 2010^ said:It is commonly recognized that the genesis of exercise hyperpnoea lies within the central nervous system and is modulated by peripheral reflexes and chemical signals. The central command, which originates in the posterior hypothalamus and the midbrain, couples the activation of the skeletal and respiratory muscles and also elicits an early HR increase [10]
As others have pointed out, some people have tachycardia, rather than chronotropic incompetence although perhaps there could be an overall flattening of the heart rate curve.
One hypothesis is that the low performance of patients is due to low cardiac output, but this can only explain things for a minority of patients. It is true that patients with congenital heart disorders tend to have exercise intolerance that can be explained due to limited cardiac output (one person I know was cured after they finally convinced a surgeon to operate).
I will say that in the recently published 2 day CPET study by Nelson et al, they tested various hypotheses relating to heart rate increases and also HR during the recovery phase and found no differences between patients and controls (not yet published data).
Do you think the lowered HR increase is secondary to impaired muscle tissue metabolism, perhaps by dysautonomic failure to recruit sufficient muscle fibers or perhaps by dysautonomic failure to dilate the arterioles supplying the muscle fibers with oxygen?
My hypothesis which is different from that of chronotropic incompetence or dysautonomic failure, rather the muscle fibres are not able to put out the same level of force for a given level of stimulation on the second day of the CPET, at around the ventilatory threshold, leading to more muscle fibres being recruited, and a greatly increased sense of effort as a result, leading to patients stopping the test earlier.
The reason for this is uncertain. We do know that the issue is unlikely to be due to the mitochondria themselves.
Natelson, Vermeulen and Wong propose some issue with oxygen transport to the muscle.
http://www.clinsci.org/content/97/5/603
https://translational-medicine.biomedcentral.com/articles/10.1186/1479-5876-8-93
https://www.ncbi.nlm.nih.gov/pubmed/1446478
Various researchers including certain Australians have proposed issues with pH buffering, which could also be due to K+, rather than merely lactate. Along with other metabolic abnormalities, but it is uncertain how these fit into a coherent theory.
Phair proposes reduced central drive due to the IDO/Tryptophan metabolic trap. I disagree with this as it is inconsistent with what we know about exercise physiology and the findings so far, but we'd need EMG measurements to completely dismiss that hypothesis.
An aside, in neuromuscular diseases, there is a disporportionate increase in VO2 without workrate increases: https://www.ncbi.nlm.nih.gov/pubmed/6684223
Last edited:
My guess is its impossible. How do you force the impossible?why didnt they force the mecfs to go to the "predicted heart rate max" ?
what would happen ?
Yeah, that was my first question about it. Those two plot lines are just a bit too neat and clean.If the x axis is change in heart rate rather than actual heart rate why isn't there much more scatter?
Pyrrhus
Established Member (Voting Rights)
My hypothesis which is different from that of chronotropic incompetence or dysautonomic failure, rather the muscle fibres are not able to put out the same level of force for a given level of stimulation on the second day of the CPET, at around the ventilatory threshold, leading to more muscle fibres being recruited, and a greatly increased sense of effort as a result, leading to patients stopping the test earlier.
The reason for this is uncertain. We do know that the issue is unlikely to be due to the mitochondria themselves.
Thank you so much, @Snow Leopard , for the intriguing crash course in exercise physiology. I still have a lot to learn about this subject.
Your hypothesis makes perfect sense. And I see why an EMG would be able to confirm or refute your hypothesis. But I wouldn’t say that your hypothesis is necessarily inconsistent with chronotropic incompetence, dysautonomic failure, or mitochondrial problems.
For contrast, I’ll share my current understanding/hypothesis:
1) Impaired functioning of hypothalamus and brainstem lead to the following two dysautonomic failures:
—a) Failure to dilate the arterioles supplying the exercising muscles with oxygen, which leads to:
——i) A lower anaerobic threshold
——ii) Overstressed mitochondria ejecting large amounts of H+ ions into the extracellular space
——iii) Increase in micro-tears in muscle fibers, caused by the increased oxidative stress
—b) Failure to increase the heart rate (HR) appropriately in response to exertion, resulting in:
——i) Chronotropic incompetence OR
——ii) POTS
2) In the 1-3 days after exercise, there is temporary mitochondrial dysfunction as the mitochondria heal, leading to:
—a) Reduction in force generated by muscle contraction, leading to:
——i) Recruitment of additional muscle fibers on day 2 of CPET
——ii) Increased perception of effort
Please let me know which of the above are inconsistent with what is known about exercise physiology, or inconsistent with the literature!
Thanks again.
Edited: removed reference to P2X receptors and delayed onset muscle soreness
Last edited:
Snow Leopard
Senior Member (Voting Rights)
1) Impaired functioning of hypothalamus and brainstem lead to the following two dysautonomic failures:
—a) Failure to dilate the arterioles supplying the exercising muscles with oxygen
Is this the problem though?
Dilation of arteries leads to reduced blood pressure and heart rate due to increased blood flow.
Note that the post exercise hypotension noted in healthy people is likely associated with prolonged dilation of arteries (1-2 days) and proportionally more blood being distributed to the periphery. It is suggested that this occurs due to the withdrawal of the sympathetic tone (which was increased during exercise), leading to lower than usual levels of catecholamines.
During recovery, a response to this vasodilation is increased sympathetic activity and reduced vagal tone.
Peripherally, the cells in blood vessels try to balance their response to catecholamines by altering their expression of alpha vs beta adrenergic receptors for example. But peripheral vasodilation in response to exercise is primarily driven by local chemical factors.
https://www.ncbi.nlm.nih.gov/pubmed/8282635
https://www.nature.com/articles/1001377
https://courses.washington.edu/conj/heart/arterioles2011.htm
It's kind of ironic that there are two opposing theories one is the "persistent arousal" hypothesis by Wyller and others which proposes increased sympathetic drive (and all the associated psychobabble of "stress" etc), whereas Phair's hypothesis involves the opposite, attenuated sympathetic drive...
Last edited:
Pyrrhus
Established Member (Voting Rights)
Peripherally, the cells in blood vessels try to balance their response to catecholamines by altering their expression of alpha vs beta adrenergic receptors for example. But peripheral vasodilation in response to exercise is primarily driven by local chemical factors.
Thank you for the clarification. It sounds like one might have to posit anti-adrenergic antibodies, not some central mechanism, if one wanted to posit failure of arteriole dilation then. (Assuming it’s consistent with observations on blood pressure during exertion.)
I’m curious- you haven’t mentioned David Systrom’s work, what is your take on his work? As I understand it, his work concerns the failure of central veins to constrict appropriately during exercise, which could be due to dysautonomia.
It's kind of ironic that there are two opposing theories one is the "persistent arousal" hypothesis by Wyller and others which proposes increased sympathetic drive (and all the associated psychobabble of "stress" etc), whereas Phair's hypothesis involves the opposite, attenuated sympathetic drive...
I have always felt that it seems more like a parasympathetic problem, there is something wrong with the rest phase. Normal resting doesn't work and I have always felt that, even when mild, a PEM cascade seemed to start the moment I started to rest.
Snow Leopard
Senior Member (Voting Rights)
I’m curious- you haven’t mentioned David Systrom’s work, what is your take on his work? As I understand it, his work concerns the failure of central veins to constrict appropriately during exercise, which could be due to dysautonomia.
I haven't looked into it in any depth, he believes exercise intolerance in POTS patients is due to pre-load failure.
But I don't see why the central veins would fail to constrict with the peripheral veins also failing to dilate at the same time?
Snow Leopard
Senior Member (Voting Rights)
it seems, that inclining (uphill) walking has a huge impact on heart rate (thats what i understand)
has this been tested, ever ?
how mecfs / controls rate in incline exercise?
The main issue with inclines is simply that it takes substantially more power to climb than walking on a flat plane. Otherwise there is nothing particularly special.
A graded CPET on a treadmill can simulate specific gradients by requiring a specific amount of power.