Good afternoon everyone,
I am enclosing below in summary the main points of our review and the questions that the model could answer about these diseases.
Many people wonder why EBV, the Epstein-Barr virus, and not another virus, is the main trigger of the symptomatic picture of ME/CFS and Long COVID, when there are other factors, such as different infections, metal intoxication, among others. In the following lines, I will explain the fundamentals of this phenomenon, summarizing the essential points of the review article we recently published.
The common denominator of all the triggering factors of both diseases is their capacity to immunosuppress. Any intracellular infection, whether bacterial, viral or parasitic, that is not controlled, leads to immunosuppression due to chronic exposure to antigens. Usually, the ability to control an infection is due to genetic factors, particularly the HLA genes (I and II). These genes encode proteins called human leukocyte antigen (HLA), which are essential for distinguishing self from non-self in our body. If this "alert" does not function properly, the immune system cannot efficiently eliminate pathogens. HLAs are like a scanner that allows us to identify things that are not from the body and respond to them. If this scanner is not working properly and does not recognize something that is bad, it would prevent your immune system from eliminating it and therefore prevent it from moving freely through your body without being eliminated.
In the context of ME/CFS, there are two possible scenarios: the individual may have a direct genetic predisposition to EBV, or they may lose control over EBV due to an underlying immune deficiency caused by another infection or exposure to chemicals or metals. If it is another infection, we would also be talking about a "genetic weakness" (HLA) to those specific pathogens.
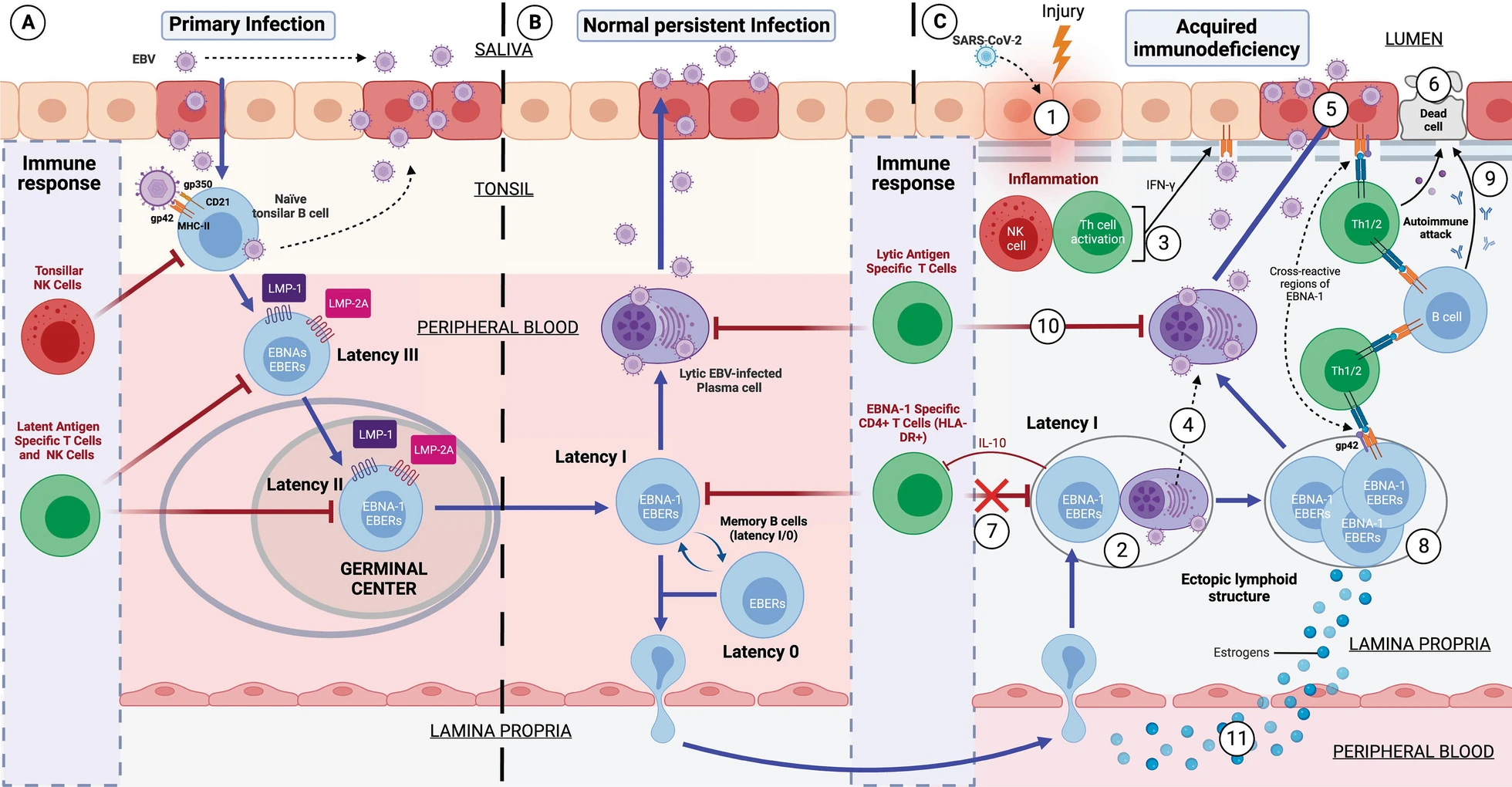
If the individual is genetically susceptible to EBV, it is because he or she has old HLA-II haplotypes, to which EBV has co-evolved. This is mainly because EBV infects by binding to HLA-II proteins in cells. This virus is cunning: when it infects a cell, it introduces its DNA into it latently without generating new virions, avoiding detection by the immune system and using B cells as "Trojan horses".
While 95% of the world's population has EBV, only a minority develop problems. This is where the "weak" HLA-II haplotypes against EBV come into play again.
Although it is said that the majority of the population does not have problems with this virus, this is not entirely true. Think that this virus takes advantage of every time you become immunosuppressed for any reason. An example is its brother herpes labialis, that every time that person is immunosuppressed for any reason, it takes advantage and infects more cells, making the lesion visible on the lips. But every time the immune system recovers from the first event that immunosuppressed him, this virus is controlled again and the lesion disappears. This is exactly the same thing that happens in healthy people who are able to control EBV.
The main difference between these healthy people and ME/CFS patients who have problems with EBV, is that by having strong HLA-II haplotypes against EBV they are able to recognize all EBV latency types and control them. In contrast, patients with EBV ME/CFS, having weak haplotypes against EBV, their TCD4 cells are not able to recognize EBV latency I cells. The rest of the latency types are able to be recognized since they are controlled by TCD8 cells, as they would not have weak HLA-I haplotypes against EBV.
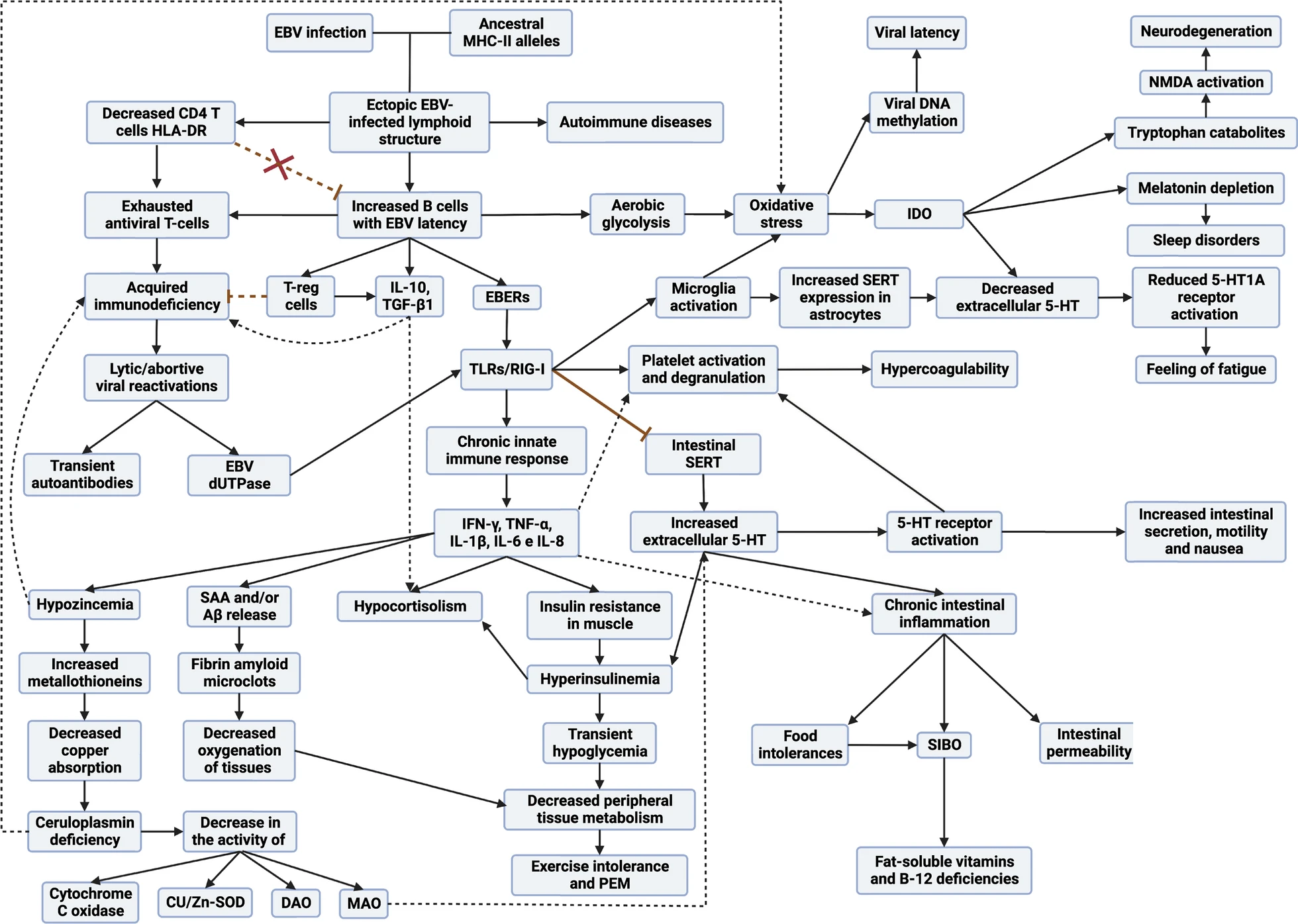
Let us explain this better. TCD4 cells recognize antigens presented on HLA-II proteins and TCD8 cells recognize antigens presented on HLA-I proteins. Normally, any intracellular infection is controlled by TCD8 cells because the infected cells present on the HLA-I proteins of their membrane the antigens of the pathogen inside. On the other hand, HLA-II proteins are found mostly on antigen-presenting cells that take antigens from outside the cell and present them on their HLA-II proteins so that they are recognized by T-CD4 cells. So, we would think that EBV, being an intracellular pathogen, its antigens should always be presented on the HLA-I proteins of the infected cell. This is not always the case; this virus has evolved to evade this system by generating latency. One of the evasion mechanisms to remain unrecognized is to prevent a latency antigen, called EBNA-1, from being presented on HLA-I proteins and from being presented on HLA-II proteins. This is of utmost importance for the virus because it prevents, for example, latency I cells (only expressing EBNA-1 from the virus) from being recognized by TCD8 cells. On the other hand, the rest of the latency cells (II and III) and lytic cells without would be recognized and eliminated by CD8 T cells. So we would think that no one would be able to control the latency I cells if they evade CD8 T cells. Here our immune system is also intelligent and this is where CD4 T cells play the differential role between healthy and sick people with this virus. The importance of HLA-II haplotypes reappears. Those individuals with EBV-resistant HLA-II haplotypes will be able to present the EBNA-1 antigen well on latency I cells and will be recognized and eliminated by TCD4 cells. In contrast, those with EBV-weak HLA-II haplotypes will not be able to present EBNA-1 well on HLA-II proteins and therefore CD4 T cells will not recognize the latency I cells. These latency I cells, when left unchecked, will multiply and cause inflammation and damage. But every time they go to another type of latency or lytic phase they will be recognized by CD8 T cells.
So what happens to those individuals who have been infected with other pathogens (such as Long COVID) and fail to control them? Well, in the end the same thing happens. Being genetically weak against these pathogens, they also end up presenting an immunodeficiency due to chronic exposure to antigens, decreasing the effective response of CD4 T cells. As these cells are the main cells that control EBV latency I, cells with latency I end up evading the immune system and multiplying, generating the same problems as in the case of EBV MS/CFS.
Therefore, in any subtype of patient with ME/CFS and in Long COVID, they end up presenting a viral syndrome due to EBV. Once EBV latency I cells are out of control, it allows any inflammatory stimulus in any tissue to recruit leukocytes (including EBV latency cells), ultimately leading to the formation of EBV-infected ectopic lymphoid aggregates.
These formations are "ectopic" because they are outside their usual location, i.e., they do not form in primary (such as thymus and bone marrow) or secondary (such as lymph nodes and spleen) lymphoid tissues. They form transiently to cope with infection or inflammation and disappear when the stimulus is resolved.
The B cells with EBV latency use these lymphoid aggregates to their advantage, since as there is antigen presentation in these structures, they take advantage of it to pass from latency to lytic phase, generating foci of viral reactivations in these inflamed tissues. This causes that, although the initial inflammatory stimulus that provoked the formation of these aggregates has been resolved, these aggregates remain continuously in these tissues due to another inflammatory stimulus due to the molecules released by the cells with latency, as well as the viral reactivations. This would occur mainly in the mucous membranes of our organism but can occur in different tissues and would be the basis for the development of autoimmune diseases.
These autoimmune diseases are generated due to the presentation of cellular autoantigens from those tissues or by the presentation of viral EBNA-1 through HLA-II proteins. The antigens when presented on HLA-II proteins undergo a series of modifications that can lead to the formation of new antigens "different" from the previous ones. In addition, EBNA-1 has a sequence similar to different proteins in our body, which can confuse our immune system and generate an autoimmune response. Here again appears the implication of having weak HLA-II haplotypes against EBV, since most of the diseases associated with this virus such as multiple sclerosis, lupus, rheumatoid arthritis, Sjögren's, etc. are associated with the same old HLA-II haplotypes as those weak against EBV. So having these autoimmune diseases implies that they do not control well these cells with EBV latency and therefore are responsible for the development of their autoimmunity.
Why do women have a higher prevalence of ME/CFS, Long COVID and autoimmune diseases?
Hormones play a crucial role. Estrogens, in particular, affect the CD4/CD8 T-cell ratio by reducing it, increase B-cell longevity, enhance antibody release and amplify the expression of HLA-II proteins. Under normal conditions, this increase in antibody levels in women enhances their resistance to viral infections. However, under pathological circumstances, this hormonal balance leads to a prolonged survival of B cells and a decrease in CD4 T cells, in addition to intensifying the expression of HLA-II. This situation leads to increased vulnerability to EBV due to increased survival of virus-transformed B cells and increased expression of HLA-II, which facilitates infection of a greater number of cells. In addition, elevated HLA-II expression can lead to a more robust presentation of cellular autoantigens or viral antigens that, after undergoing post-translational changes, generate neoantigens. These can activate autoreactive cells. Therefore, both the increased survival of transformed B cells and the increased antigenic presentation generated by the increased expression of HLA-II by estradiol may favor an increase in the presentation of both self and foreign antigens during an infectious process, and those abnormal plasma cells that produce autoantibodies survive longer, which consequently increases the likelihood of women developing autoimmune diseases or even cancer.
On the male side, testosterone modulates the immune system by promoting CD4 Th1 (antiviral) response and CD8 T-cell activation, while inhibiting NK cell response and HLA-II expression. Since antigen-presenting cells are essential for the differentiation of CD4 T cells toward Th1 or Th2, based on the cytokines they release, sex hormones may influence this differentiation. Women, having higher levels of antibodies (Th2 response), show a more efficient response to extracellular infections. However, against intracellular pathogens, their immune system may not be as efficient as the male immune system, which benefits from a stronger Th1 cellular response to fight virus-infected cells.
Does this model explain the overactive yet ineffective immune response?
Yes, this model describes a situation in which the body's immune system is working excessively, but inefficiently, against the EBV virus:
1. Deficient adaptive response: CD4 T cells, are not correctly recognizing and dealing with EBV latency I cells. This leads to more infected cells circulating and, at the same time, to a fatigue or exhaustion of the T cells, reducing their ability to fight the virus.
2. Over-activation of innate immunity: Because of this failure of the adaptive response, another part of the immune system, called innate immunity, becomes over-activated because it continually detects that there is an infection but that it cannot be resolved by adaptive immunity. This results in the constant production of inflammatory substances that, instead of helping, can generate more problems and maintain chronic inflammation.
3. Imbalance in immune responses: The body tends to favor an immune response (known as Th2) that is not best suited to fight this type of infection. This is due in part to the production of a substance called IL-6, which redirects the body's defensive response towards the production of antibodies instead of an antiviral cellular response (Th1). The increased Th2 response favors the latency and lytic cycles of EBV by activating more B cells.
Does this model explain viral reactivations and that of other latent pathogens?
Yes, the model explains that, due to decreased activation and function of cytotoxic CD4 T cells, there is a loss of immune surveillance over latent infections of other pathogens. These CD4 T cells are necessary to control latent or lytic phase cells of pathogens such as Parvovirus B19, EBV, cytomegalovirus and other herpesviruses. As a result, increased viral reactivation will be observed, especially in individuals with a higher degree of immunodeficiency.
Does this model explain how there could be increased metal intoxication in these patients?
If there is increased expression of MTs due to elevated intracellular zinc levels, as described in the model, those MTs will be busy binding and regulating zinc and, potentially, copper. As a result, there would be fewer MTs available to bind and detoxify heavy metals that may be present. This could lead to an accumulation of unregulated and potentially toxic heavy metals (such as cadmium and mercury) in the body.
Does this model explain the increased oxidative stress in these diseases?
Yes, the model explains the increased oxidative stress. Infected ectopic lymphoid structures trigger inflammatory responses by releasing certain viral components. This activation induces the release of proinflammatory cytokines, which, in turn, promote excessive production of reactive oxygen species, leading to marked oxidative stress. In addition, perturbation in the homeostasis of certain metals contributes to the disruption of intracellular antioxidant responses. Specifically, there is evidence that an antioxidant enzyme (superoxide dismutase) is affected by altered copper and zinc concentration. Briefly, the model describes how the combination of chronic inflammatory responses, together with imbalances in the homeostasis of certain metals and the persistent release of proinflammatory agents, culminates in a significant increase in oxidative and nitrosative stress in the body.
Does this model explain exercise intolerance and post-exertional malaise?
Yes, this model explains exercise intolerance and post-exertional distress in the context of persistent EBV infection and its metabolic, immunological and neurophysiological interactions. The following is a breakdown of how the model addresses this phenomenon:
1. Metabolic alterations: the model describes how EBV infection can lead to insulin resistance through elevated IFN-γ production. This resistance, accompanied by compensatory hyperinsulinemia, can lead to transient hypoglycemia and decreased peripheral tissue metabolism. These factors contribute to exercise intolerance, as muscles are unable to obtain the necessary energy efficiently, resulting in early fatigue.
2. Alterations in cardiovascular function: High levels of serotonin and activation of certain receptors, such as TLR3 and TLR2, can alter cardiovascular function, affecting blood distribution and the body's ability to meet oxygen demands during exercise.
3. Compromised thermoregulation: The body's ability to dissipate heat generated during exercise may be impaired, which could lead to overheating.
4. Oxidative stress: Chronic infection with EBV generates constant oxidative stress, which can impair mitochondrial function and reduce the ability of muscle tissue to generate energy efficiently. This oxidative stress exacerbated during exercise can lead to cell damage and muscle fatigue.
5. Alterations in respiratory function: Respiratory function may be impaired, limiting adequate oxygenation during exercise and contributing to fatigue.
6. Systemic inflammation: Activation of certain receptors, release of proinflammatory cytokines and other mechanisms associated with chronic infection can generate systemic inflammation. This inflammation can negatively affect the body's ability to recover after exercise, contributing to post-exertional malaise.
7. Alterations in neurological function: Metabolic changes and systemic inflammation can have an impact on the central nervous system. Reduced serotonin availability and other alterations may contribute to feelings of fatigue and lethargy.
 EBV as a link: This review article suggests that EBV may be a catalyst, inducing similar symptoms in Long COVID and Myalgic Encephalomyelitis, and orchestrating far-reaching immune challenges.
EBV as a link: This review article suggests that EBV may be a catalyst, inducing similar symptoms in Long COVID and Myalgic Encephalomyelitis, and orchestrating far-reaching immune challenges. Immunodeficiency and Ectopic Lymphoid Aggregates: One of the most intriguing and alarming findings regarding EBV is its ability to induce the formation of structures called ectopic lymphoid aggregates in tissues. These structures are not benign; in fact, they can be potent instigators of inflammatory responses that disrupt normal tissue function. Why does this occur? This review suggests that in individuals with certain genetic characteristics - specifically those with "weak" HLA-II haplotypes against EBV - this virus can become more easily established, leading to the formation of these aggregates. Most worryingly, these aggregates not only cause inflammation, but may also contribute to a form of acquired immunodeficiency, further weakening the body's defenses and even developing autoimmune diseases.
Immunodeficiency and Ectopic Lymphoid Aggregates: One of the most intriguing and alarming findings regarding EBV is its ability to induce the formation of structures called ectopic lymphoid aggregates in tissues. These structures are not benign; in fact, they can be potent instigators of inflammatory responses that disrupt normal tissue function. Why does this occur? This review suggests that in individuals with certain genetic characteristics - specifically those with "weak" HLA-II haplotypes against EBV - this virus can become more easily established, leading to the formation of these aggregates. Most worryingly, these aggregates not only cause inflammation, but may also contribute to a form of acquired immunodeficiency, further weakening the body's defenses and even developing autoimmune diseases. Consequences:
Consequences: Sex Differences: The role of gender differences is critical in affecting EBV interaction and symptom manifestation. Biological sex may influence the interaction with EBV. Estrogens in women increase B-cell survival and antibody release, but may also amplify risks with EBV, potentially promoting autoimmune conditions.
Sex Differences: The role of gender differences is critical in affecting EBV interaction and symptom manifestation. Biological sex may influence the interaction with EBV. Estrogens in women increase B-cell survival and antibody release, but may also amplify risks with EBV, potentially promoting autoimmune conditions. Treatments that could improve or worsen symptoms:
Treatments that could improve or worsen symptoms: