Im going to try to explain what this study found.
First, we need to be familiar with the structure of antibody. An antibody is an immunoglobulin (Ig) that can come in several
classes (G, A, M, D and E). This is the prototypical antibody:
Notice the red part. This is called the
heavy chain. The blue part is called the
light chain. Each light chain is connected to a heavy chain via disulphide bonds and the heavy chains themselves are connected via disulphide bonds, giving a macromolecule with
two heavy chains and two light chains.
An antibody recognises and covalently binds to a specific "thing". This "thing" is called an
antigen and can be many things: proteins, LPS, even hormones. In order for the body to recognise a wide variety of antigens, it makes sense to have many different antibodies. This is achieved by having a
constant region, that determines the common characteristics of all antibodies (for example, the class), and a
variable region. This variable region is the one that binds to antigen, and comes in many different shapes.
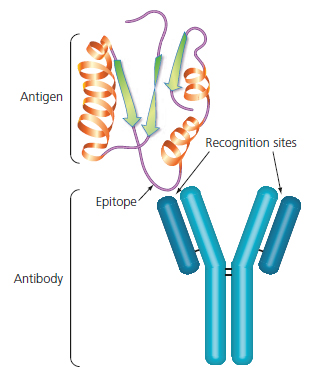
This is how an antibody binds. In this case, the antigen is a protein, and the small part that the antibody recognises is called
epitope. An epitope is a part of the antigen, and the same antigen can have different epitopes. For example a big protein has different epitopes, each one corresponding to some different amino acid sequence.
It is in the best interest of the body to recognise a wide variety of different antigens, in order to have a broad spectrum of defence, but it is impractical to have thousands or tens of thousands of genes, each coding for a different variable region.
So how does the body generate the observed antibody diversity? By a complex process called
somatic recombination. In this process, different genes are joined together to form variable regions. Because of the many different ways the genes can be joined, the diversity increases exponentially while using comparatively few genes.
This is how somatic recombination works. For the Variable part of the heavy chain, a particular
Variable gene is chosen. This gene is combined with a
Diversity gene, and the two combine with a
Join gene. This is called V(D)J recombination, and the VDJ sequence is finally joined to a
Constant gene, that encodes the constant region.
For the light chain, only Variable and Join genes combine to form the Variable part, therefore the light chain has less diversity.
Once the heavy chain and light chain are transcribed and translated, the protein products join to form a functioning antibody.
The antibody producing cells are called B cells, and each B cell produces only one kind of antibody (though exceptions occur). Over the lifetime of a B cell however, it can
class switch. Changing the Constant region of the heavy chain to a different class.
A B cell is first inactive, generating antibody and using it as a membrane bound receptor, the
B Cell Receptor (BCR). When the membrane receptor recognises and binds to antigen, the B cell becomes activated. It processes the encountered antigen and presents it to
T helper cells. If the T cell also recognises it, it "authorises" the B cell to activate fully.
The B cell rapidly replicates, migrates to the blood and tissues, and starts secreting the antibody. This process of rapid replication upon activation is called
clonal expansion, and most of the clonal expanded B cells produce the same antibody, although there are special mechanisms to subtly change the variable region of a few B cells to generate more potent antibodies.
The image above is an example of clonal expansion.
Now, what did Lipkin find? He found a particular protein elevated in the blood. This protein is called IGHV3-23, this stands for
Immune Globulin Heavy chain Variable 3 -23. This gene is one of the Variable genes that we talked bout earlier.
The reasoning is that because this protein is elevated in blood, some B cell has become activated and is producing large quantities of one particular antibody that contains IGHV3-23, therefore generating large amounts of this IGHV3-23.
B cell activation is suggestive of chronic infection or autoimmunity. Which one is it? I have no idea
")