Jonathan Edwards
Senior Member (Voting Rights)
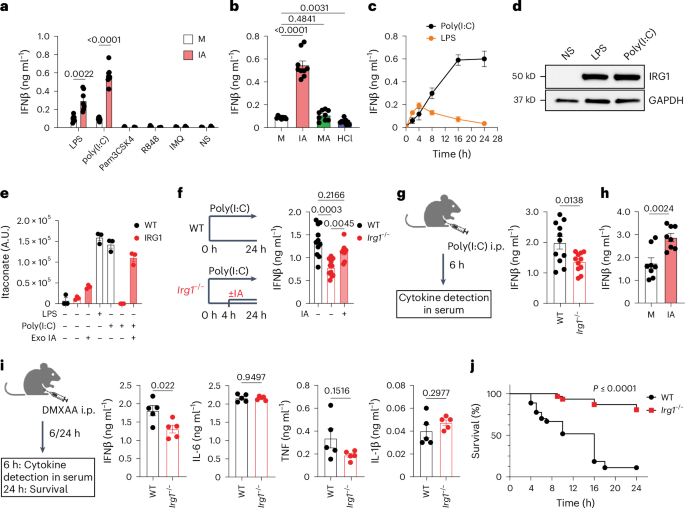
I apologise for trying to make sense of all the threads around DecodeME with another thread but I see interest in collecting thoughts on interferons as candidates. This follows on from recent discussion on the Genetics OLFM4 thread but it goes back much further.
"Interferon' was discovered by two people in 1957, one being a close family friend who died in his forties when I was a child (Alick Isaacs, to a child a wonderfully amusing and kind 'uncle'). It turned out to be several things and we are probably mostly interested in alpha and beta (type 1 Ifs) and gamma (type 2). My understanding is that although these are separate molecules and have different repertoires they often up-regulate expression of the same proteins and pathways.
Interest in ME/CFS goes back to the observation that alpha interferon therapy causes fatigue. Robert Phair picked up on alpha interferon as a key mediator in his itaconate shunt idea. @jnmaciuch and I have been discussing the merits of alpha and gamma interferons for some months. Gamma interferon seemed plausible as a T cell signal that might make more sense than some of the cytokines like TNF or IL6 as part of a persisting abnormal immune response. Now @jnmaciuch has pointed out that we might use alpha interferon as a way to tie in several gene variant hits in DecodeME. At least some of those might link to gamma interferon too, but maybe not all.
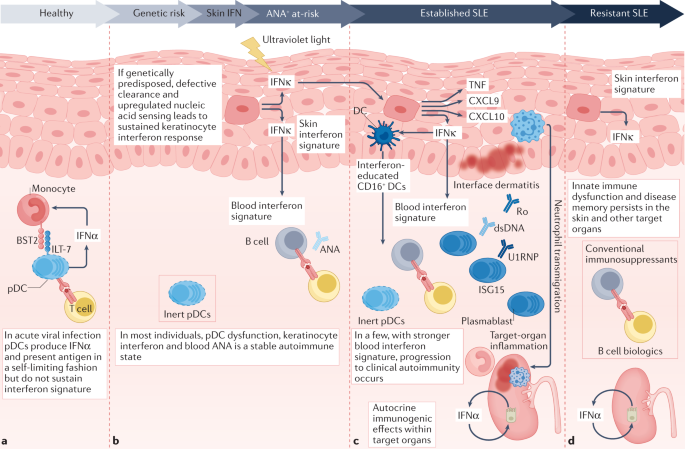
My sense is that discussion of pooled knowledge on these molecules and pathways would be a good place to start in trying to build ideas around DecodeME results. Key questions are going to be 1. which tissues might specifically be hosting local, maybe hidden, interferon-based signalling and how that might relate to symptoms 2. how to understand time courses for inappropriate interferon signalling over both short and long term in ways that would make biological sense and fit symptomatology.
I think that is about enough for an introductory post but I will add a few quick thoughts.
1. I am coming around to the idea that gamma interferon may be involved early on in disease triggering but that alpha (?+beta) may well take over long term.
2. For me the problem with alpha is how the body 'learns' to overproduce or over-respond with the long term dynamics of ME/CFS but the increased focus on brain (or maybe neurons) from DecodeME might be relevant there. (Learning to make more gamma is the standard job of T cells.)
3. How best do we tie the DecodeME hits together into a story and what awkward questions would that raise.
"Interferon' was discovered by two people in 1957, one being a close family friend who died in his forties when I was a child (Alick Isaacs, to a child a wonderfully amusing and kind 'uncle'). It turned out to be several things and we are probably mostly interested in alpha and beta (type 1 Ifs) and gamma (type 2). My understanding is that although these are separate molecules and have different repertoires they often up-regulate expression of the same proteins and pathways.
Interest in ME/CFS goes back to the observation that alpha interferon therapy causes fatigue. Robert Phair picked up on alpha interferon as a key mediator in his itaconate shunt idea. @jnmaciuch and I have been discussing the merits of alpha and gamma interferons for some months. Gamma interferon seemed plausible as a T cell signal that might make more sense than some of the cytokines like TNF or IL6 as part of a persisting abnormal immune response. Now @jnmaciuch has pointed out that we might use alpha interferon as a way to tie in several gene variant hits in DecodeME. At least some of those might link to gamma interferon too, but maybe not all.
My sense is that discussion of pooled knowledge on these molecules and pathways would be a good place to start in trying to build ideas around DecodeME results. Key questions are going to be 1. which tissues might specifically be hosting local, maybe hidden, interferon-based signalling and how that might relate to symptoms 2. how to understand time courses for inappropriate interferon signalling over both short and long term in ways that would make biological sense and fit symptomatology.
I think that is about enough for an introductory post but I will add a few quick thoughts.
1. I am coming around to the idea that gamma interferon may be involved early on in disease triggering but that alpha (?+beta) may well take over long term.
2. For me the problem with alpha is how the body 'learns' to overproduce or over-respond with the long term dynamics of ME/CFS but the increased focus on brain (or maybe neurons) from DecodeME might be relevant there. (Learning to make more gamma is the standard job of T cells.)
3. How best do we tie the DecodeME hits together into a story and what awkward questions would that raise.