Jonathan Edwards
Senior Member (Voting Rights)
I like that idea @jnmaciuch . I am boating again but will add some thorts later.
Very interesting! How would we test the hypothesis that type I interferons are involved in ME/CFS?Looking through the candidate list again, I think there's an interesting thread that ties several of the top genes together, which is regulation of type I (alpha/beta) interferon signaling.
OLFM4
No directionality info from eQTLs.
PEBP1
TRIM38 (type I interferons are one of the main products of TLR and cGAS-STRING/viral RNA sensing)
KLHL20
E3-ubiquitin ligases are typically associated with degradation of interferon, but it might not be so cut and dry.
ZNFX1
I'll note that though this is an interferon-stimulated gene, it's activity as a dsRNA sensory would also trigger interferon.
And interestingly, PRDX6, a cousin of PRDX5--which is inhibited by itaconate to allow type I interferon production in macrophages
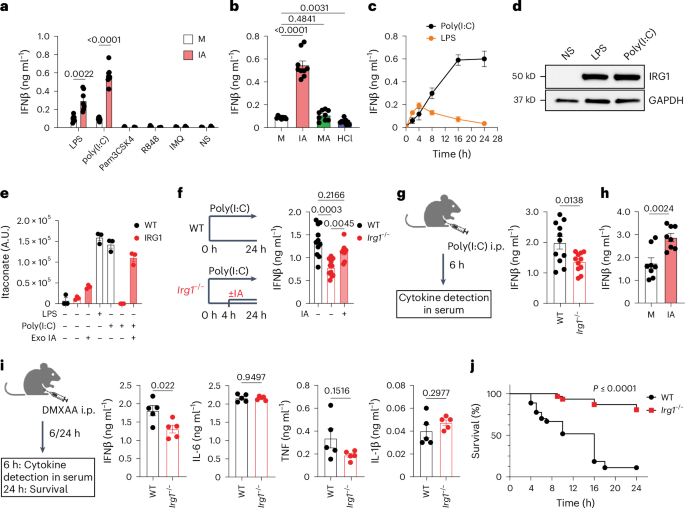
Itaconate modulates immune responses via inhibition of peroxiredoxin 5 - Nature Metabolism
Itaconate is shown to non-covalently inhibit the antioxidant enzyme peroxiredoxin 5 in macrophages, thereby modulating the production of mitochondrial peroxide and enhancing the type I interferon production.www.nature.com
The BTNs could also be part of the story via their regulatory effect on TLRs. Plus TLR/type I interferon signaling utilizes MAPK signaling, which potentially implicates ZNF322 (increased) and SUDS3 (increased).Plus PTGIS (decreased expression), since prostaglandins are known to inhibit interferon signaling in some contexts.
Just throwing things at a wall to see what sticks based on the assumption that at least some of these genes are actually affected by the identified SNPs. The directionality of most of these (where eQTL data is provided) seems to generally be in the direction of enhanced interferon in ME/CFS (either increased expression in genes that are positive regulators or decreased in genes that are negative regulators), so that would be some weak evidence against the idea that these SNPs are driven by [edit: poorer protection in ME/CFS cases] against viral infections.
[Edit: and acknowledging that I’m probably biased here since I already think there’s a good case for type I interferons in ME/CFS]
I unfortunately can't give details about projects involving data from another research group, but I can say that I'm already on the case and have reasons to be encouraged so farVery interesting! How would we test the hypothesis that type I interferons are involved in ME/CFS?
") (I know, the most annoying response to hear from a researcher!)
(I know, the most annoying response to hear from a researcher!) One option is to run high-sensitivity assays for interferon on blood samples from pwME in active PEM, which probably would probably be quite difficult to arrange but is potentially doable for a team of able-bodied researchers who can drive around for home visits.
They’re not standard assays, unfortunately, though if it’s possible to find enough people that can travel while in PEM then sample collection could just happen as normal. If it gets funded by one of the charities maybe they could be convinced to include funds to pay for a driver to make the trek as painless as possible. Or at least one could dream.If they're standard assays, it might not be unsurmountable for moderately affected people with access to a general hospital.
This makes intuitive sense to me but that is probably meaningless.It could potentially be a combination of both as well, if interferon production starts from a trigger in the tissue and eventually increases in concentration until it reaches the blood during PEM.
One option is to run high-sensitivity assays for interferon on blood samples from pwME in active PEM, which probably would probably be quite difficult to arrange but is potentially doable for a team of able-bodied researchers who can drive around for home visits.
I know LIINC have a lot of tissue samples from pwLC, perhaps they have some of the tissues you theorise are affected? But of course access is a problem and probably not possible to know which test subjects experience PEM.The second option, which could potentially provide more mechanistic insight but would be much more technically involved, is to collect (and possibly culture) cells from the tissues you think are affected and measure interferon before and after some stimulation.
Looking through the candidate list again, I think there's an interesting thread that ties several of the top genes together, which is regulation of type I (alpha/beta) interferon signaling.
OLFM4
No directionality info from eQTLs.
PEBP1
TRIM38 (type I interferons are one of the main products of TLR and cGAS-STRING/viral RNA sensing)
KLHL20
E3-ubiquitin ligases are typically associated with degradation of interferon, but it might not be so cut and dry.
ZNFX1
I'll note that though this is an interferon-stimulated gene, it's activity as a dsRNA sensory would also trigger interferon.
And interestingly, PRDX6, a cousin of PRDX5--which is inhibited by itaconate to allow type I interferon production in macrophages
Itaconate modulates immune responses via inhibition of peroxiredoxin 5 - Nature Metabolism
Itaconate is shown to non-covalently inhibit the antioxidant enzyme peroxiredoxin 5 in macrophages, thereby modulating the production of mitochondrial peroxide and enhancing the type I interferon production.
The BTNs could also be part of the story via their regulatory effect on TLRs. Plus TLR/type I interferon signaling utilizes MAPK signaling, which potentially implicates ZNF322 (increased) and SUDS3 (increased).Plus PTGIS (decreased expression), since prostaglandins are known to inhibit interferon signaling in some contexts.
Just throwing things at a wall to see what sticks based on the assumption that at least some of these genes are actually affected by the identified SNPs. The directionality of most of these (where eQTL data is provided) seems to generally be in the direction of enhanced interferon in ME/CFS (either increased expression in genes that are positive regulators or decreased in genes that are negative regulators), so that would be some weak evidence against the idea that these SNPs are driven by [edit: poorer protection in ME/CFS cases] against viral infections.
[Edit: and acknowledging that I’m probably biased here since I already think there’s a good case for type I interferons in ME/CFS]
From Entrez: This gene encodes a member of the phosphatidylethanolamine-binding family of proteins and has been shown to modulate multiple signaling pathways, including the MAP kinase (MAPK), NF-kappa B, and glycogen synthase kinase-3 (GSK-3) signaling pathways. The encoded protein can be further processed to form a smaller cleavage product, hippocampal cholinergic neurostimulating peptide (HCNP), which may be involved in neural development. This gene has been implicated in numerous human cancers and may act as a metastasis suppressor gene. Multiple pseudogenes of this gene have been identified in the genome. [provided by RefSeq, Jul 2015]
From Uniprot: Substrate-specific adapter of a BCR (BTB-CUL3-RBX1) E3 ubiquitin-protein ligase complex involved in interferon response and anterograde Golgi to endosome transport. The BCR(KLHL20) E3 ubiquitin ligase complex mediates the ubiquitination of DAPK1, leading to its degradation by the proteasome, thereby acting as a negative regulator of apoptosis 1. The BCR(KLHL20) E3 ubiquitin ligase complex also specifically mediates 'Lys-33'-linked ubiquitination 2. Involved in anterograde Golgi to endosome transport by mediating 'Lys-33'-linked ubiquitination of CORO7, promoting interaction between CORO7 and EPS15, thereby facilitating actin polymerization and post-Golgi trafficking 3. Also acts as a regulator of endothelial migration during angiogenesis by controlling the activation of Rho GTPases. The BCR(KLHL20) E3 ubiquitin ligase complex acts as a regulator of neurite outgrowth by mediating ubiquitination and degradation of PDZ-RhoGEF/ARHGEF11 4. In case of tumor, the BCR(KLHL20) E3 ubiquitin ligase complex is involved in tumor hypoxia: following hypoxia, the BCR(KLHL20)complex mediates ubiquitination and degradation of PML, potentiating HIF-1 signaling and cancer progression
That’s the drop of blood study, yeah? Definitely worth reaching out, thanks for the reminder.Chris Armstrong's team are/were doing a study that involved sampling people in active PEM with home visits iirc, so he might be a good person to consult on the logistics of that.
No worries, I believe so. I found these two links from a quick google. I think there is more info on S4ME but I am brain fogged right now!That’s the drop of blood study, yeah? Definitely worth reaching out, thanks for the reminder.

 www.omf.ngo
www.omf.ngo
www.omf.ngo
www.omf.ngo
That’s definitely something I’m interested in—part of what drew me to the type I interferon response is that it is nearly universal across cell types, and thus could potentially explain PEM triggered by both muscle activity and cognitive activity for some pwME. It seemed to fit quite well since we already know that mtDNA release triggered in muscle during exercise elicits a mild type I interferon response in healthy people, and interferon therapy side effects are remarkably similar to PEM symptoms.For example is there a inteferon response happening within the neurons (or glia) themselves or would something like that be happening elsewhere and affecting neurons through cell interactions etc.
When extremely severe, I experienced both those symptoms. (Memory loss more as in inability to form new memories, like I have entire weeks where a month after I could remember absolutely nothing from them but a “feeling”.)memory loss or psychosis seen in either of those conditions.
I hadn’t previously heard of pwME experiencing any psychosis—that’s very useful to know. That’s probably because the people who experience it are the least able to communicate online. And, I’m sure, many pwME might feel that it will be used against them by psychosomatic proponents.When extremely severe, I experienced both those symptoms. (Memory loss more as in inability to form new memories, like I have entire weeks where a month after I could remember absolutely nothing from them but a “feeling”.)
Yes. It’s actually somewhat commonly talked about on private groups for severe people. But rarely brought up publicly lest get institutionalised. Perhaps it’s also just a natural consequence of having very little visual or auditory stimulation like your brain starts compensating by making stuff up.I hadn’t previously heard of pwME experiencing any psychosis—that’s very useful to know. That’s probably because the people who experience it are the least able to communicate online. And, I’m sure, many pwME might feel that it will be used against them by psychosomatic proponents.
There’s a Norwegian team called the Moser Group (by the nobel winners) that works on how memory is formed.I wonder if in dementia it’s actually a two-part phenomenon: something preventing new memories from being made (perhaps something influenced by interferon) and something that destroys neurons harboring older memories (which doesn’t happen in ME/CFS, as far as it aware, though I could be wrong).
 www.ntnu.edu
www.ntnu.edu
Very valuable, thank you for sharing. It seems I have some chatting to do with a member of my former department that studies neurolupusYes. It’s actually somewhat commonly talked about on private groups for severe people. But rarely brought up publicly lest get institutionalised. Perhaps it’s also just a natural consequence of having very little visual or auditory stimulation like your brain starts compensating by making stuff up.
It’s very possible, though I did have something similar to what @Yann04 described when I was more moderate—stretches of months where I knew that something happened but the details aren’t so clear later on. I think I just didn’t connect it to “memory loss” until this discussion (and it was probably milder than what severe pwME describe). I definitely had plenty of stimulation even though I was housebound. It might be worthwhile to have a more detailed survey on that across severitiesThere’s a Norwegian team called the Moser Group (by the nobel winners) that works on how memory is formed.
Moser Group - Kavli Institute for Systems Neuroscience - NTNU
Kavli Institute's Space and memory Group
See e.g. this:
It’s way above my understanding, but I remember a news article where they explained that memories are stored in sequences of events that are determined based on events that stand out. These events are like tying knots on a string with pearls that keeps groups of pearls together (as distinct memories). The knots (specific activation of neurons) are always unique.
If the brain has very little stimuli, maybe there are very few knots so memories aren’t formed properly? Pure speculation by me.
That’s perhaps a grand word for what I’m doingI think @hotblack was theorizing a similar “cracks in the system under duress” idea in the context of brain metabolism on another thread
")
It's exactly the same as what any of us researchers and grad students are doing!That’s perhaps a grand word for what I’m doing
That's definitely the question, and it's what I've been tinkering at with my hypothesis. If I'm actually onto something re: type I interferons and not just seeing what I want to see, the question becomes "what could go wrong in interferon signaling that sustains a disease state over many years, but doesn't result in out-of-control damage a la interferonopathies or inflammatory diseases that involve interferon?"Really interesting thoughts on potential things which could tie the identified contributors together. So if these are all factors which would increase the chance of a runaway process, or spin little cogs in a way which makes it more likely, what’s the actual process or cog?
Makes sense. Thanks for all the explanation. An always ready and waiting pathway that is perhaps a little too ready and always sounds particularly appealing for ME/CFS. Could it also be that this state 2 is doing something new and different it doesn’t in healthy people, rather than just a how much it’s turned up or suppressed?The resolution of infection involves switching over from 1 to 2. My hunch, given the lack of inflammatory damage that you'd expect from constantly being in state 1, is that it's more likely to be a problem fully switching over to state 2 or keeping state 2 suppressed enough