Jacob Richter
Established Member (Voting Rights)
Abstract
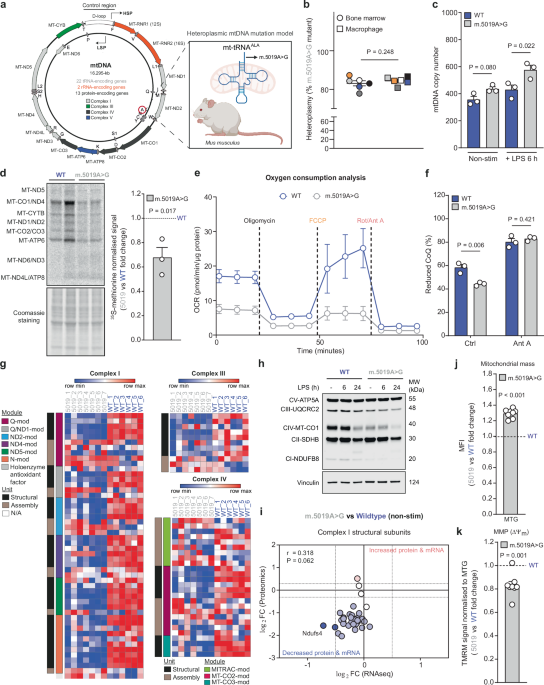
Impaired mitochondrial bioenergetics in macrophages promotes hyperinflammatory cytokine responses, but whether inherited mtDNA mutations drive similar phenotypes is unknown. Here, we profiled macrophages harbouring a heteroplasmic mitochondrial tRNAAla mutation (m.5019A>G) to address this question. These macrophages exhibit combined respiratory chain defects, reduced oxidative phosphorylation, disrupted cristae architecture, and compensatory metabolic adaptations in central carbon metabolism. Upon inflammatory activation, m.5019A>G macrophages produce elevated type I interferon (IFN), while exhibiting reduced pro-inflammatory cytokines and oxylipins. Mechanistically, suppression of pro-IL-1β and COX2 requires autocrine IFN-β signalling. IFN-β induction is biphasic: an early TLR4-IRF3 driven phase, and a later response involving mitochondrial nucleic acids and the cGAS-STING pathway. In vivo, lipopolysaccharide (LPS) challenge of m.5019A>G mice results in elevated type I IFN signalling and exacerbated sickness behaviour. These findings reveal that a pathogenic mtDNA mutation promotes an imbalanced innate immune response, which has potential implications for the progression of pathology in mtDNA disease patients.
 www.nature.com
www.nature.com
Impaired mitochondrial bioenergetics in macrophages promotes hyperinflammatory cytokine responses, but whether inherited mtDNA mutations drive similar phenotypes is unknown. Here, we profiled macrophages harbouring a heteroplasmic mitochondrial tRNAAla mutation (m.5019A>G) to address this question. These macrophages exhibit combined respiratory chain defects, reduced oxidative phosphorylation, disrupted cristae architecture, and compensatory metabolic adaptations in central carbon metabolism. Upon inflammatory activation, m.5019A>G macrophages produce elevated type I interferon (IFN), while exhibiting reduced pro-inflammatory cytokines and oxylipins. Mechanistically, suppression of pro-IL-1β and COX2 requires autocrine IFN-β signalling. IFN-β induction is biphasic: an early TLR4-IRF3 driven phase, and a later response involving mitochondrial nucleic acids and the cGAS-STING pathway. In vivo, lipopolysaccharide (LPS) challenge of m.5019A>G mice results in elevated type I IFN signalling and exacerbated sickness behaviour. These findings reveal that a pathogenic mtDNA mutation promotes an imbalanced innate immune response, which has potential implications for the progression of pathology in mtDNA disease patients.
An inherited mitochondrial DNA mutation remodels inflammatory cytokine responses in macrophages and in vivo in mice - Nature Communications
Inherited mitochondrial DNA mutations can result in diverse clinical phenotypes. Here, the authors characterise a heteroplasmic tRNAAla mutation (m.5019A>G) in mice and demonstrate that macrophages carrying this mutation display altered function and metabolism in vitro, along with increased type...
www.nature.com